Le malattie rare rappresentano un vasto e complesso universo medico, caratterizzato da un numero elevato di patologie (oltre 6.000 in tutto il mondo) che, pur singolarmente poco diffuse, nel loro insieme colpiscono milioni di persone. In Italia, si stima che circa 2 milioni di individui vivano con una di queste condizioni, spesso genetiche e croniche, che richiedono un’attenzione e un impegno crescenti da parte della comunità scientifica, medica e sociale. La Giornata mondiale delle malattie rare, che si celebra il 29 febbraio (o il 28 negli anni non bisestili), è un momento cruciale per accendere i riflettori su queste patologie, portandole fuori dal "cono d'ombra" di ignoranza e indifferenza in cui troppo spesso rimangono confinate.

La Sindrome di Wiskott-Aldrich: Un Esempio di Malattia Genetica Rara
Una delle malattie genetiche rare che illustra la gravità e le sfide associate a queste condizioni è la Sindrome di Wiskott-Aldrich (WAS). Si tratta di una patologia del sistema immunitario, ereditaria e legata al cromosoma X, che può avere conseguenze potenzialmente letali, anche per infezioni comuni come un banale raffreddore. La WAS è causata da mutazioni nel gene WAS, che codifica per una proteina essenziale per il corretto funzionamento dei globuli bianchi, in particolare per la loro mobilità e interazione con altre cellule. Questa disfunzione immunitaria si traduce in una maggiore suscettibilità alle infezioni, ma anche in un aumento del rischio di sviluppare malattie autoimmuni e tumori.
I sintomi della WAS possono manifestarsi fin dalla prima infanzia e includono eczema grave, trombocitopenia (basso numero di piastrine, che causa sanguinamenti anomali e petecchie) e un’aumentata incidenza di infezioni ricorrenti. La gravità della malattia è molto variabile, ma in assenza di trattamento, la prognosi può essere infausta.
Wiskott Aldrich: nella terapia genica una nuova speranza
La Storia di Aidan: Dalla Diagnosi Difficile alla Speranza della Terapia Genica
La storia di Aidan, un nome di fantasia utilizzato per proteggere la privacy, è emblematica delle difficoltà che molte famiglie affrontano nella diagnosi e nella gestione delle malattie rare. Nato in Albania nel 2003, Aidan ha iniziato a manifestare sintomi legati alla WAS fin dai sei mesi di vita: febbre continua, difficoltà respiratorie e infezioni frequenti erano la sua quotidianità. Nonostante la sofferenza, Aidan ha dimostrato una notevole forza di volontà, riuscendo a frequentare la scuola e a gestire autonomamente le sue terapie. La sua famiglia, di umili origini contadine, ha compiuto enormi sacrifici per garantirgli le cure necessarie, con il fratello maggiore che si è trasferito in Italia per lavorare e contribuire alle spese mediche.
La diagnosi della WAS per Aidan non è stata semplice. In Albania, i medici faticavano a identificarne la causa. È stata l'intraprendenza della sorella maggiore, che osservando attentamente i sintomi e ricercando informazioni online, a portare all'identificazione della Sindrome di Wiskott-Aldrich. Questa scoperta ha aperto la strada a un percorso di speranza.
Grazie all'aiuto di un'associazione umanitaria, la famiglia di Aidan è riuscita a raggiungere l'Italia, dove, dopo essere transitati per Novara e Brescia, sono giunti all'Istituto San Raffaele Telethon per la terapia genica a Milano. In Italia è arrivata la diagnosi definitiva e, purtroppo, anche la constatazione di danni fisici preesistenti: l'occhio destro, completamente cieco, è stato asportato, mentre il sinistro, compromesso, è stato sottoposto a trapianto di cornea.
L'Innovazione Terapeutica: La Terapia Genica di Telethon
L'Istituto San Raffaele era all'avanguardia nella ricerca e nello sviluppo di trattamenti innovativi per le malattie genetiche rare. In particolare, era in corso un trattamento sperimentale di terapia genica per la WAS. Sebbene le spese per la terapia e il soggiorno in Italia fossero coperte da Fondazione Telethon, la famiglia ha dovuto affrontare ulteriori sacrifici, vendendo i propri animali per potersi permettere il lungo soggiorno necessario per il trattamento. Durante questo periodo, Aidan, allora di dieci anni, ha vissuto un misto di agitazione e speranza, consapevole che quel viaggio in Italia rappresentava un'opportunità per cambiare il corso della sua vita.
La terapia genica è una metodologia rivoluzionaria che mira a correggere la causa genetica della malattia. Nel caso della WAS, il principio è quello di prelevare le cellule staminali ematopoietiche del paziente, modificarle geneticamente in laboratorio per correggere il difetto nel gene WAS, e reinfonderle nel paziente. Queste cellule corrette sono in grado di produrre globuli bianchi sani, ripristinando così la funzionalità del sistema immunitario.

Il 22 aprile 2013, Aidan è stato trattato con successo. Il percorso terapeutico, sebbene impegnativo e segnato dalla separazione temporanea dalla famiglia, ha portato a risultati straordinari. Oggi, Aidan sta magnificamente bene, avendo superato non solo le problematiche legate alla malattia, ma anche le sfide dell'adolescenza. Vive a Pisa con la sua famiglia e continua a tornare periodicamente a Milano per controlli, considerando l'ospedale come un luogo di ritorno a "casa", dove incontrare le persone che gli hanno donato una seconda vita.
La storia di Aidan è un potente esempio di come l'innovazione scientifica, unita all'impegno di fondazioni come Telethon e alla determinazione dei pazienti e delle loro famiglie, possa offrire speranza e migliorare significativamente la qualità della vita di chi è affetto da malattie genetiche rare. L'Agenzia europea per i medicinali ha riconosciuto l'importanza di questo programma, selezionando Telethon per il suo progetto pilota di accelerazione terapeutica, un segnale positivo per l'intero settore delle malattie rare.
L'Adrenoleucodistrofia Legata all'X (X-ALD): Una Malattia Degenerativa Complessa
Un altro esempio significativo di malattia genetica rara è l'Adrenoleucodistrofia Legata all'X (X-ALD), una patologia degenerativa grave che colpisce il sistema nervoso e le ghiandole surrenali. La X-ALD è causata da mutazioni nel gene ABCD1, situato sul cromosoma X, che è responsabile della produzione di una proteina coinvolta nell'eliminazione degli acidi grassi saturi a catena molto lunga (VLCFA). L'accumulo di questi VLCFA nel tessuto nervoso, in particolare nelle cellule della glia che producono mielina, innesca una risposta immunitaria che porta alla demielinizzazione, ovvero alla progressiva distruzione della mielina che riveste le fibre nervose. Questo processo patologico compromette la capacità dei neuroni di comunicare tra loro e con il resto del corpo.
Manifestazioni Cliniche e Fenotipi della X-ALD
La X-ALD si manifesta clinicamente in tre fenotipi principali, con varianti e sfumature che rendono la diagnosi e la gestione particolarmente complesse:
- Adrenoleucodistrofia Cerebrale Legata all'X (X-CALD): Questa è la forma più comune e aggressiva, che colpisce prevalentemente bambini e adolescenti (tra i 3 e i 12 anni). I sintomi neurologici sono severi e progressivamente ingravescenti. Spesso, i primi segni sono legati a una ridotta attività surrenalica, ma evolvono rapidamente in gravi difficoltà di apprendimento, disfagia (difficoltà a deglutire), sordità, perdita della vista, convulsioni e atassia (perdita di coordinazione motoria), potendo portare fino a uno stato neurovegetativo.
- Adrenomieloneuropatia (AMN): Tipicamente esordisce in età adulta (tra i 20 e i 30 anni) con sintomi a progressione lenta. Questi possono includere rigidità degli arti, debolezza e dolore a mani e piedi, spasmi muscolari e problemi urinari. In alcuni casi, può evolvere verso uno stato vegetativo. Possono manifestarsi anche disturbi del comportamento e cognitivi. Le donne portatrici, sebbene solitamente asintomatiche o con forme lievi, possono sviluppare segni di AMN ad esordio tardivo (oltre i 40 anni).
- Adrenoleucodistrofia con Insufficienza Corticosurrenalica (AI) isolata: Circa il 70% dei maschi con X-ALD presenta insufficienza corticosurrenalica, che può essere il primo e unico sintomo per anni, precedendo di decenni la comparsa dei sintomi neurologici. Questa condizione, simile alla malattia di Addison, è causata dall'accumulo di VLCFA nelle ghiandole surrenali e si manifesta con affaticamento cronico, nausea, perdita di peso, ipotensione e ipoglicemia.
Esiste anche una forma neonatale (NALD), ma questa va distinta dalla X-ALD e rientra nello spettro dei disturbi della biogenesi dei perossisomi.
Diagnosi e Trattamento della X-ALD
La diagnosi di X-ALD si basa sull'osservazione clinica dei sintomi e viene confermata da specifici test biochimici e genetici. Il dosaggio dei VLCFA nel plasma è un elemento chiave, così come l'analisi genetica per identificare le mutazioni nel gene ABCD1.
Il trattamento della X-ALD è complesso e dipende dal fenotipo, dalla gravità dei sintomi e dalla progressione della malattia. Per l'insufficienza corticosurrenalica, si ricorre alla terapia sostitutiva con glucocorticoidi e mineralcorticoidi, mimando i livelli fisiologici di secrezione ormonale. La terapia fisica è utile per trattare la spasticità, soprattutto nella forma AMN.
Un aspetto fondamentale del trattamento, valido per tutte le forme di adrenoleucodistrofia, è la terapia dietetica di supporto. Questa si basa su una dieta a basso contenuto di VLCFA e sull'uso di alimenti a fini medici speciali, costituiti da miscele di oli in grado di abbassare i livelli di questi acidi grassi.
L'unica cura potenzialmente risolutiva per la forma cerebrale (X-CALD) è il trapianto di midollo osseo. Questo intervento è raccomandato per i pazienti, soprattutto bambini, con un donatore compatibile e, crucialmente, prima che la demielinizzazione cerebrale sia estesa e i sintomi neurologici gravi. La diagnosi precoce è quindi determinante per permettere al paziente di beneficiare di questa opzione terapeutica, che altrimenti rischia di non avere esiti favorevoli.
Wiskott Aldrich: nella terapia genica una nuova speranza
La Sindrome di Ehlers-Danlos Miopatica (mEDS): Una Sfida Diagnostica e Terapeutica
La Sindrome di Ehlers-Danlos (EDS) è un gruppo eterogeneo di disturbi ereditari del tessuto connettivo, caratterizzati da ipermobilità articolare, lassità dei legamenti, fragilità cutanea e tissutale. Tra le numerose varianti, la Sindrome di Ehlers-Danlos Miopatica (mEDS) rappresenta una sfida diagnostica e terapeutica particolare. La storia di Aiden (un altro nome di fantasia) illustra bene queste difficoltà.
Aiden è stato diagnosticato a 7 anni e 5 mesi con mEDS, dopo un lungo percorso segnato da sintomi non specifici e difficili da collegare. Fin dalla nascita, i medici avevano notato un basso tono muscolare, un soffio al cuore e fianchi "flaccidi". Nei primi anni di vita, Aiden ha lottato con reflusso gastroesofageo, problemi digestivi, disturbi del sonno, ritardo nella crescita e ritardi nel raggiungimento delle tappe fondamentali dello sviluppo.
Sintomi e Diagnosi della mEDS
Uno degli episodi più drammatici nella vita di Aiden è stato il peggioramento della deviazione oculare, diagnosticata inizialmente come strabismo. L'intervento chirurgico, previsto per essere di un'ora, si è protratto per oltre due ore, poiché il chirurgo ha scoperto che le orbite di Aiden erano deformate, una condizione ben più grave del previsto. Questo ha richiesto ulteriori otto interventi chirurgici agli occhi e un intervento addominale prima dei dieci anni.
Negli anni successivi, Aiden ha sofferto di lussazioni articolari dolorose, rotture tendinee, emicranie debilitanti e complicazioni digestive sempre più gravi. Nonostante i sospetti di EDS, la diagnosi è rimasta elusiva. Aiden non soddisfaceva i criteri standard per la diagnosi di EDS ipermobile (hEDS), in particolare per quanto riguarda il punteggio sulla scala Beighton di ipermobilità articolare, che non era sufficientemente elevato.
Solo dopo il sesto intervento agli occhi, la famiglia è stata indirizzata al reparto di genetica. Anche lì, la genetista ha notato che l'ipermobilità non era abbastanza pronunciata per una diagnosi di hEDS, ma ha suggerito la possibilità di altre forme, avvertendo che si sapeva poco di alcune di esse. La ricerca personale della madre ha portato alla scoperta della mEDS, una forma meno comune e meno compresa dell'EDS, che ha dato un senso retroattivo a molti dei sintomi di Aiden.
La Sfida dell'Advocacy e della Gestione Multidisciplinare
La gestione della mEDS, come per molte altre malattie rare, richiede un approccio multidisciplinare, coinvolgendo specialisti in cardiologia, neurologia, ortopedia, gastroenterologia, fisioterapia e genetica. L'ostacolo maggiore, come sottolinea la madre di Aiden, è "collegare i sintomi e accettare che non sempre esiste una risposta chiara e univoca. Non sempre esiste un percorso chiaro."
In queste situazioni, diventa fondamentale l'advocacy, ovvero il ruolo attivo dei pazienti e delle loro famiglie nel cercare risposte, nel sensibilizzare e nel fare pressione per una maggiore comprensione e ricerca. Essere "la ruota che cigola", "il disco rotto", il paziente che "sono di nuovo qui" è spesso necessario per ottenere l'attenzione e le cure adeguate.
Aiden ha affrontato ogni sfida con un coraggio ammirevole, stupendo con il suo atteggiamento e la sua determinazione. La sua storia, e quella di innumerevoli altri pazienti affetti da malattie genetiche rare, sottolinea l'importanza di non arrendersi, di continuare a fare domande e di sostenere la ricerca e la diffusione di informazioni.
La Malattia di Addison e l'Insufficienza Surrenalica
Sebbene non sia strettamente una malattia genetica nel senso di ereditarietà diretta per ogni individuo, la Malattia di Addison (o insufficienza surrenalica primaria cronica, o iposurrenalismo) è una condizione endocrina che può essere causata da fattori autoimmuni, infettivi o, in alcuni casi, da predisposizioni genetiche che influenzano il sistema immunitario. La forma di insufficienza surrenalica legata all'accumulo di VLCFA nell'adrenoleucodistrofia, come discusso precedentemente, è un esempio di come le patologie genetiche possano influenzare le ghiandole surrenali.
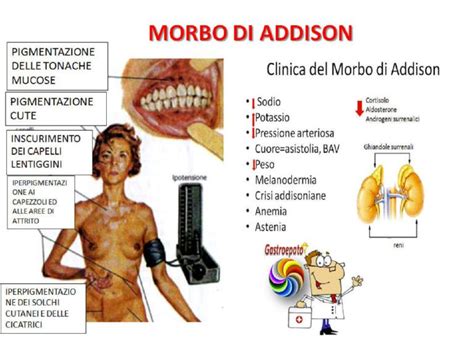
La Malattia di Addison si manifesta tipicamente in modo insidioso, con sintomi non specifici che possono essere confusi con altre patologie più comuni. Tra i sintomi più frequenti vi sono: affaticamento cronico, perdita di forze, malessere generale, perdita di peso, nausea, anoressia (che nei bambini si traduce in ritardo della crescita), dolori muscolari e articolari.
Un segno cardinale, spesso presente, è l'iperpigmentazione cutanea e delle mucose, che si manifesta con un inscurimento della pelle, in particolare nelle pieghe palmari, sulle nocche, sulle cicatrici, nella mucosa orale e nei siti di frizione. Sintomi come ipotensione posturale (calo della pressione sanguigna alzandosi in piedi) e ipoglicemia (bassi livelli di zucchero nel sangue) tendono a comparire più tardivamente. I pazienti possono manifestare un desiderio intenso di ingerire sale. Sono inoltre frequenti manifestazioni come la vitiligine e l'alopecia areata.
La malattia causa un deficit di deidroepiandrosterone (DHEA), un ormone steroideo che può portare a sindromi specifiche nelle donne, come la perdita di peli ascellari/pubici, l'assenza di pubarca nei bambini, la riduzione della libido e la secchezza cutanea.
Diagnosi e Gestione dell'Insufficienza Surrenalica
La diagnosi di Malattia di Addison viene confermata mediante test biochimici mirati. È essenziale misurare, al mattino, i livelli sierici di cortisolo e dell'ormone adrenocorticotropo (ACTH) nel plasma. Nell'insufficienza surrenalica primaria, l'ACTH plasmatico è tipicamente molto elevato, mentre i livelli di cortisolo mattutino sono bassi. Un test di stimolazione con ACTH esogeno è utile per confermare la diagnosi: nei soggetti sani, il cortisolo aumenta significativamente dopo la somministrazione dell'ormone, mentre nei pazienti con Addison la risposta è assente o molto ridotta.
È fondamentale escludere l'insufficienza surrenalica secondaria (causata da problemi all'ipofisi) e altre condizioni che possono presentare un quadro clinico simile, come tumori ipofisari, tubercolosi, infezioni fungine o malattie infiltrative.
La gestione della Malattia di Addison richiede un approccio multidisciplinare e una presa in carico a vita. Il trattamento principale consiste nella terapia sostitutiva con glucocorticoidi, solitamente idrocortisone orale, somministrato in dosi frazionate per mimare la secrezione fisiologica del cortisolo. È necessario anche il fludrocortisone orale per rimpiazzare gli ormoni mineralcorticoidi. La sostituzione con DHEA è opzionale e valutata caso per caso.
Il monitoraggio dei livelli di glucocorticoidi è cruciale, specialmente durante periodi di stress (malattie, interventi chirurgici), per prevenire crisi addisoniane (AAI), che possono essere pericolose per la vita. Il dosaggio dell'idrocortisone viene calibrato in base alla risposta clinica del paziente, considerando il suo benessere generale e l'eventuale presenza di segni di sovra- o sottodosaggio. L'attività della renina plasmatica può essere monitorata per ottimizzare il dosaggio del fludrocortisone. Nei bambini, è fondamentale monitorare la crescita e lo sviluppo.
Sebbene non esista un trattamento risolutivo che curi la causa sottostante, con una terapia appropriata e un attento monitoraggio, le aspettative di vita per i pazienti con Malattia di Addison non sono ridotte.
Conclusioni Implicite: La Necessità di Ricerca, Sensibilizzazione e Supporto
Le storie di Aidan e dei pazienti affetti da Sindrome di Wiskott-Aldrich, adrenoleucodistrofia e sindrome di Ehlers-Danlos, pur riguardando patologie diverse, evidenziano temi comuni nel panorama delle malattie rare. La complessità diagnostica, la necessità di competenze specialistiche multidisciplinari, i sacrifici economici e personali affrontati dalle famiglie, e l'importanza cruciale della ricerca scientifica per lo sviluppo di terapie innovative.
La Giornata mondiale delle malattie rare serve a ricordare che dietro ogni patologia rara c'è una persona, una famiglia, una storia che merita attenzione e speranza. L'impegno delle reti di associazioni, il lavoro di Fondazione Telethon e di altre organizzazioni, e i progressi della medicina genica e di precisione stanno gradualmente portando queste condizioni fuori dall'ombra, offrendo nuove prospettive terapeutiche e migliorando la qualità della vita dei pazienti. Continuare a investire nella ricerca, promuovere la sensibilizzazione e garantire un accesso equo alle cure sono passi fondamentali per costruire un futuro più inclusivo e supportivo per tutti coloro che vivono con una malattia rara.